Density Functional Theory Study of the Mechanisms and Stereochemistry of the Rh(I)-Catalyzed Intramolecular [3+2] Cycloadditions of 1-Ene- and 1-Yne-Vinylcyclopropanes.
Abstract
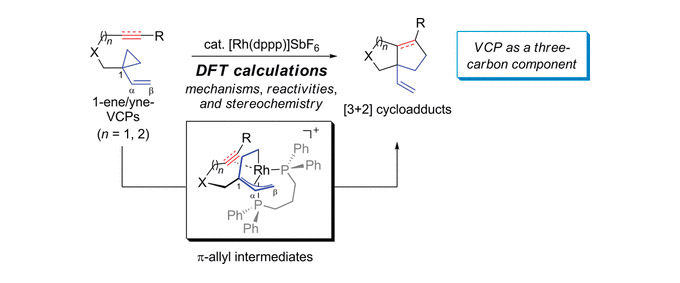
The mechanisms, structures of all stationary points involved, and kinetic and thermodynamic parameters of the Rh(I)-catalyzed intramolecular [3+2] cycloaddition reactions of 1-ene- and 1-yne-vinylcyclopropanes (1-ene-VCPs and 1-yne-VCPs) have been investigated using density functional theory (DFT) calculations. The computational results showed that the [3+2] reactions of 1-ene/yne-VCPs studied here occur through a catalytic cycle of substrate−catalyst complex formation, cyclopropane cleavage, alkene/alkyne insertion, and reductive elimination. Alkene/alkyne insertion is the rate- and stereoselectivity-determining step of these multistep [3+2] cycloadditions. The experimentally observed high reactivity of 1-yne-VCPs compared to 1-ene-VCPs is well rationalized by the differences of steric effects in the alkyne/alkene insertion transition states. DFT calculations unveiled that the relative orientation of the tethers in the 1-ene/yne-VCPs plays a key role in controlling the stereochemistry of the [3+2] cycloadducts. In addition, DFT calculation results are used to explain why, in some cases, the formation of the β-hydride elimination byproduct can compete with the [3+2] pathway.